| Présentation du logiciel | Sommaire | Tutorial, partie 2 |
Cette partie présente, sur un exemple, le principe de l'extraction et de l'analyse d'un spectre EXAFS à l'aide du logiciel LASE. Seules les opérations essentielles sont présentées ici ; pour des explications détaillées sur chacune des options et sur leur fonctionnement, reportez-vous aux parties suivantes. Le composé choisi pour cet exemple est le complexe [Fe(OPhMe2)4)]NEt4. Le spectre d'absorption a été mesuré en transmission sur la ligne EXAFS III du LURE, sur un échantillon en poudre, au seuil K du fer ; différentes études indiquent que, dans ce complexe, le fer occupe le centre d'un tétraèdre d'atomes d'oxygène. Pour davantage d'informations à propos de ce complexe et de l'analyse de son spectre d'absorption X, voir Travaux préliminaires à l'étude par spectroscopie d'absorption X de complexes modèles du site actif de la nitrile-hydratase, Emmanuel CURIS, rapport de stage de DEA, 1996.
Cette partie est organisée de la façon suivante :
Le signal EXAFS est, par définition, l'ensemble des oscillations normalisées du coefficient d'absorption atomique après le seuil d'absorption. L'environnement local de l'atome absorbeur est à l'origine de ces oscillations : il est donc possible d'en déduire des informations structurales. Cette déduction se fait en essayant de reproduire au mieux les oscillations expérimentales par un modèle théorique adapté. Cette modélisation est particulièrement simple pour les oscillations dues à la première couche de voisins.
Le traitement d'un spectre expérimental, donnant le coefficient d'absorption total en fonction de l'énergie du photon incident, nécessite donc l'extraction des oscillations d'origine structurale (signal EXAFS), éventuellement la séparation des différentes contributions structurales à ces oscillations, la création d'un modèle structural « grossier » permettant les calculs théoriques et la recherche des paramètres structuraux permettant de décrire au mieux les oscillations expérimentales à partir de ce modèle structural.
La première étape repose sur la définition des oscillations EXAFS, khi(k), à partir du coefficient d'absorption total, µ : µ = µm + µ0 (1 + khi(k)) donc
où µm représente la contribution des atomes autres que l'absorbeur à l'absorption de l'échantillon et µ0 est l'absorption de l'absorbeur supposé seul dans l'univers ; E est l'énergie du photon incident et k est le nombre d'onde de l'électron émis :

E0 étant l'énergie de seuil. L'extraction de khi(k) se fait donc en modélisant µm et µ0.
Dans le cadre de l'exemple choisi, nous nous limiterons à la modélisation de la contribution des premiers voisins du fer (« première couche »), soit les quatre oxygènes liés au fer. Cette contribution est isolée à l'aide de la transformation de Fourier.
Lors d'une expérience en transmission sur la ligne EXAFS III, les données enregistrées par le logiciel de pilotage (Acquvisu) sont l'énergie du photon incident (exprimée en eV), l'intensité du faisceau incident I0 (en nombre de coups) et l'intensité du faisceau transmis I1 (en nombre de coups).
Pour récupérer ces données et calculer le coefficient d'absorption total, il faut utiliser l'option Extraire le signal brut..., dans le menu Absorption.
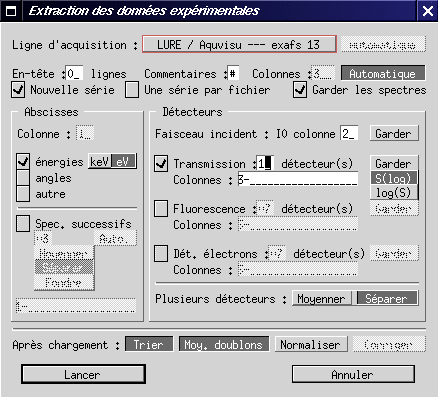
L'appel de cette fonction ouvre la boîte de dialogue ci-dessus, qui permet de préciser le mode d'acquisition (transmission ou fluorescence), le logiciel utilisé pour le pilotage et diverses options permettant un premier traitement au vol des spectres chargés. Ces options ne seront pas détaillées ici ; pour charger le fichier expérimental du complexe étudié, il faut indiquer des données enregistrées en transmission, sans multi-détecteur, sur la ligne EXAFS III, comme sur la figure ci-dessus. Le fichier à charger est fe_phen.dat, dans le dossier Exemples. Pour davantage d'informations, suivre ce lien.
Une fois le fichier chargé, le spectre d'absorption doit apparaître dans la fenêtre principale du programme.
Comme l'indique la formule définissant les oscillations EXAFS, il faut pour extraire le signal modéliser les deux termes µm et µ0. La modélisation de µm se fait en une fois ; celle de µ0 utilise une procédure en trois étapes : µ0 = µ0(I) + µ0(II) + µ0(III). Le plus simple, pour mener cette opération à bien, est de choisir l'option Tout faire... dans le menu EXAFS, qui enchaîne automatiquement ces différentes étapes, réalise la normalisation et permet de paramétrer la transformation de Fourier puis la transformation de Fourier inverse (voir la partie suivante).
Lorsque cette option est appelée, toutes les fenêtres décrites dans la suite de ce texte apparaissent simultanément et s'arrangent à l'écran : la fenêtre pour µm en haut à gauche, celle pour µ0(I) en haut à droite, celle pour µ0(II) en bas à gauche, celle pour µ0(III) en bas à droite et celle pour la transformée de Fourier au centre. La moindre modification d'un paramètre dans une fenêtre est transmise dans toutes les fenêtres suivantes, ce qui permet de contrôler rapidement l'influence du paramètre modifié sur le spectre et la qualité du spectre extrait.
Il est important de bien fermer chacune des fenêtres dans l'ordre logique, c'est-à-dire en commençant par celle de µm et en finissant par celle de µ0(III). En effet, un certain nombre de paramètres internes ne sont mis à jour que lors de la fermeture de la fenêtre (propagation des barres d'erreur, de la couleur du spectre, mémorisation des paramètres pour une extraction ultérieure, création de l'historique,...).
À la fermeture de la fenêtre pour estimer µ0(III), le spectre est normalisé. Cette normalisation s'accompagne d'une disparition des effets de la pondération par kn, ce qui provoque une modification importante de la transformée de Fourier. Cet effet est normal, il suffit de redemander une pondération par kn (menu exafs, option Multiplier par k^n).

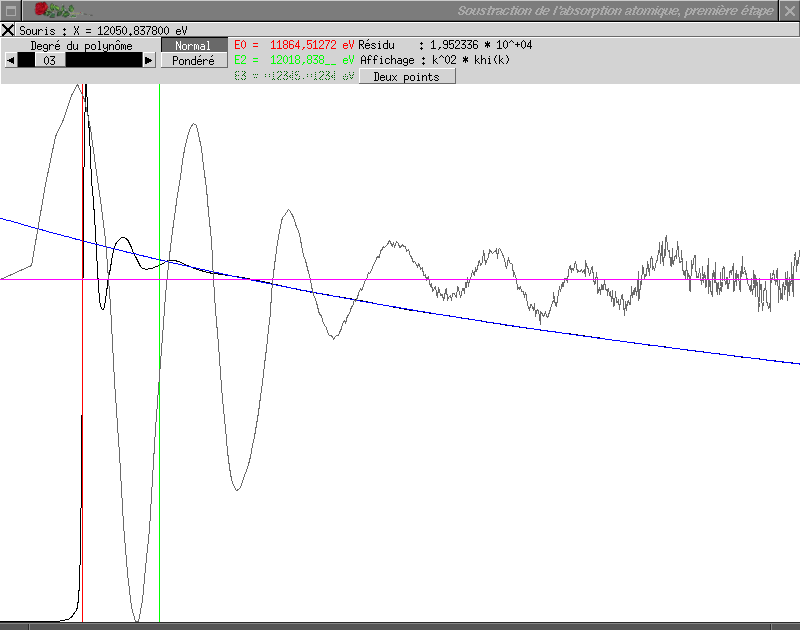
La courbe noire représente le spectre expérimental, la courbe bleue le terme µm calculé et la courbe grise le spectre résultant de la soustraction de µm, soit µ - µm. Le trait rouge vertical indique la position de l'énergie E1. Par défaut, µm est modélisé par une courbe de Victoreen, ou « victorine », d'équation y = A + Bx-3. Les paramètres A et B sont obtenus par la méthode des moindres carrés ; les points utilisés pour cela sont tous les points tels que x < E1.
La position de E1 peut être modifiée en cliquant avec le bouton gauche la nouvelle position ; il est possible d'entrer directement une valeur précise en cliquant sur la valeur numérique de E1 affichée en rouge en haut de la fenêtre.
La nature de la courbe modélisant µm peut être choisie dans le sélecteur en haut à gauche de la fenêtre. Suivez ce lien pour avoir la description détaillée de cette fonction.
Lorsque le terme µm obtenu vous satisfait, cliquez sur
le bouton de fermeture de la fenêtre et demandez le calcul du
résultat. Pour l'exemple choisi, une valeur de E1
convenable est E1 = 7085 eV.
La courbe noire représente le spectre expérimental, la courbe bleue
le terme µ0(I) calculé --- tous deux représentés en échelle
d'énergie (E, en eV) ; la courbe grise représente le spectre
EXAFS résultant de cette soustraction, en échelle des nombres d'onde
(k, en Å-1) et pondéré par kn. Le
trait rouge vertical indique la position de l'énergie de seuil,
E0, servant d'origine pour le calcul des nombres
d'onde ; le trait vert indique la position du paramètre
E2 ; le trait violet horizontal représente la position
du zéro pour le spectre EXAFS extrait (idéalement, le spectre doit
osciller autour de cet axe). L'erreur (au sens des moindres carrés)
entre le polynôme et le spectre brut, dans la partie servant à la
détermination du polynôme, est indiquée en haut à droite.
Lors de cette étape, le terme µ0(I) est modélisé par un polynôme
de degré choisi par l'utilisateur (degré 3 par défaut) ; les
coefficients du polynôme sont calculés à l'aide de la méthode des moindres
carrés à l'aide de tous les points d'énergie supérieure à E2 ;
ce polynôme est extrapolé jusqu'à l'énergie de seuil.
La position de l'énergie de seuil peut être indiquée en cliquant avec le
bouton gauche à la nouvelle position ; il est possible d'indiquer une
valeur numérique précise en cliquant sur le texte rouge
E0 = ... eV indiquant la valeur de E0
utilisée.
La position de E2 peut être indiquée en cliquant avec le
deuxième bouton à la nouvelle position ; il est possible d'indiquer une
valeur numérique précise en cliquant sur le texte vert
E2 = ... eV indiquant la valeur de E2
utilisée.
Le facteur de pondération, n, peut être modifié en cliquant sur le
texte
Affichage : k^__ * khi(k). Cette
pondération est conservée dans toute la suite du traitement, jusqu'à la
normalisation.
Lorsque le terme µ0(I) vous satisfait, cliquez sur le bouton de
fermeture de la fenêtre et demandez le calcul du résultat. Pour l'exemple
choisi, des valeurs convenables sont E0 = 7124 eV,
E2 = 7230 eV et n = 2 avec un
polynôme de degré 3.
Pour une description complète de cette fonction, suivez ce lien.
La courbe noire représente le spectre expérimental, la courbe bleue le
terme µ0(II) calculé et la courbe grise le spectre EXAFS résultant
de cette correction ; le trait violet horizontal représente la position
du zéro pour le spectre EXAFS extrait.
Le terme µ0(II) est estimé par un polynôme de degré spécifié par
l'utilisateur ; les coefficients du polynôme sont calculés par la méthode
des moindres carrés sur l'ensemble du spectre EXAFS
Lorsque le terme µ0(II) vous satisfait, cliquez sur le bouton de
fermeture de la fenêtre et demandez le calcul du résultat. Pour l'exemple
choisi, les valeurs proposées par défaut donnent des résultats convenables.
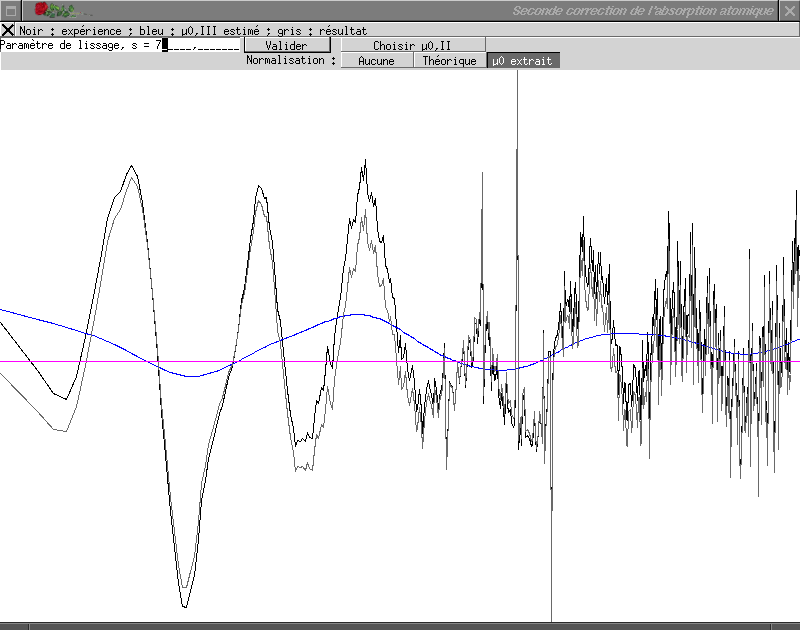
La courbe noire représente le spectre expérimental, la courbe bleue le
terme µ0(III) calculé et la courbe grise le spectre EXAFS résultant
de cette correction ; le trait violet horizontal représente la position
du zéro pour le spectre EXAFS extrait.
Le terme µ0(III) est calculé en utilisant une spline contrainte
par le paramètre s, positif (Paramètre de
lissage) ; plus ce paramètre est faible et plus le terme
µ0(III) est proche du signal brut (à la limite, pour
s = 0, ces deux grandeurs sont égales et le spectre extrait
est rigoureusement nul) ; quand le paramètre tend vers l'infini,
µ0(III) tend vers une droite. La valeur de s doit être
choisie de façon à éliminer les oscillations résiduelles à basse
fréquence ; en général les valeurs juste avant d'obtenir une droite
conviennent. La valeur de s peut être très variable d'un spectre à
l'autre et, suivant le composé, peut valoir 0,001 comme 1500. Pour entrer la
valeur, il suffit de l'indiquer après le texte
Paramètre de lissage, s = puis d'appuyer
sur Entrée.
Lorsque le terme µ0(III) vous satisfait, cliquez sur le bouton
de fermeture de la fenêtre et demandez le calcul du résultat. Pour l'exemple
choisi et avec les valeurs indiquées ci-dessus pour les étapes précédentes de
l'extraction, une valeur de 6 pour s donne de bons résultats.
Cette étape est la dernière de l'élimination de l'absorption atomique. Elle
donne
µ0 = µ0(I) + µ0(II) + µ0(III)
et µ - (µm + µ0), tous deux exprimés
dans l'échelle des nombres d'onde et pondérés par kn. Pour
obtenir khi(k), il suffit donc de diviser ces deux grandeurs. Lorsque
que l'on quitte ce module, cette division est automatiquement réalisée ;
on obtient donc directement khi(k). Il est possible d'empêcher cette
normalisation, ou de normaliser en utilisant un autre µ0 que celui
modélisé : voir la description complète
de la fonction pour davantage d'informations.
Après avoir réalisé cette extraction, le spectre obtenu n'est
pas pondéré, il faut pour ajouter une pondération utiliser la
fonction Multiplier par
kn. Pour l'exemple considéré, il faut donc appeler
l'option Multiplier par kn et demander une
pondération par k2. Il reste ensuite à isoler les
contributions à l'aide de la transformation de Fourier.
Il est maintenant possible de décomposer les oscillations EXAFS obtenues
précédemment en fonction de leur fréquence, directement reliée à la distance
parcourue par l'onde du photoélectron avant de revenir interférer sur l'atome
absorbeur ; cette décomposition en fréquence fait appel à la
transformation de Fourier, le nombre d'onde k étant la grandeur
conjuguée de cette longueur parcourue, 2R. Dans le formalisme de la réflexion
simple, chaque pic observé sur la transformée de Fourier correspond à une
« couche » de voisins (réflecteurs) situés à une même distance de
l'absorbeur ; isoler ce pic et en prendre la transformée de Fourier
inverse permet de connaître la contribution de cette couche au signal EXAFS
total. Physiquement, on n'observe plus de contribution significative pour
R > 8 Å environ ; les oscillations à plus haute
fréquence sont assimilées à du bruit : prendre la transformée de Fourier
inverse en éliminant ces fréquences permet donc d'éliminer une importante
partie du bruit.
Le signal enregistré expérimentalement n'étant pas infini, un calcul direct
de la transformée de Fourier du spectre conduit à des oscillations parasites
(oscillations de coupure). Pour amoindrir ces effets, le signal est multiplié
par une fonction qui varie doucement de 0 à 1 puis décroît vers 0, la
fonction d'apodisation ou fenêtre. Mais
cette multiplication conduit à une atténuation du signal et à la perte des
informations dans la région d'annulation de la fonction. Il faut donc trouver
un compromis entre la diminution des oscillations de coupure et l'atténuation
du signal. Le rôle de la fenêtre qui apparaît à cette étape est de paramétrer
cette fonction d'apodisation.
La courbe noire représente le spectre expérimental, la bleue représente la
fenêtre choisie, la violette représente le produit du spectre expérimental et
de la fenêtre (c'est la transformée de Fourier de cette courbe qui va être
effectivement calculée). Le trait rouge vif indique la position de
kmin, le trait vert clair celle de kmax,
le trait noir la position de la souris. Il est possible de visualiser un
aperçu de la transformée de Fourier que l'on va obtenir, en activant le bouton
Aperçu.
Le type de fenêtre utilisé (Kaiser-Bessel par défaut) est défini
par le sélecteur en haut, à gauche de la fenêtre. Suivant le type de
fenêtre, il est nécessaire de préciser de 2 à 5 paramètres pour
la définir complètement ; la fenêtre de Kaiser-Bessel est défini
par la position de kmin et kmax,
qui définissent la position du maximum, et par un paramètre qui
définit la courbure --- voir cette
partie pour davantage d'informations sur le rôle des différents
paramètres et les différents types de fenêtre.
La position de kmin peut être indiquée soit en cliquant
avec le bouton gauche de la souris la position indiquée, soit en cliquant sur
le texte affichant sa valeur.
La position de kmax peut être indiquée soit en cliquant
avec le bouton droit de la souris la position indiquée, soit en cliquant sur
le texte affichant sa valeur.
La valeur du paramètre peut être modifiée en cliquant sur le texte
affichant sa valeur.
Une fois que la fenêtre vous satisfait, il suffit de cliquer sur le bouton
de fermeture pour passer à l'étape suivante : la boîte de dialogue
ci-dessous vous permet alors d'indiquer les autres paramètres du calcul.
Pour davantage d'informations sur le rôle de ces paramètres, voyez la page
... Pour le spectre étudié en exemple, la fenêtre proposée par défaut
convient ; un calcul par la méthode normale pour R compris entre 0 et
10 Å, avec un pas de 0,02 Å, sans correction de phase ou d'amplitude
donne une transformée de Fourier utilisable dans la suite.
À la fin du calcul, la fenêtre de paramétrage de calcul de la transformée
de Fourier inverse apparaît.
Le rôle de la fenêtre qui apparaît maintenant est de définir la zone à
conserver pour le calcul de la transformée de Fourier inverse :
Cette zone est définie en multipliant la transformée de Fourier par une
fonction nulle partout où l'on veut perdre le signal et valant 1 là où l'on
veut conserver le signal, la transition étant plus ou moins brutale (il s'agit
d'un filtre passe-bande). Les fonctions utilisées sont les mêmes que les
fonctions servant de fenêtre pour le calcul de la transformée de Fourier et se
paramètrent de la même façon ; par défaut, le filtre proposé est une
fonction de Hanning asymétrique définie par quatre paramètres :
Rmin et Rmax, qui définissent les points d'annulation,
et R1 (en rouge sombre, placé par shift + bouton gauche) et
R2 (en vert foncé, placé par shift + bouton droit), qui définissent
le plateau (où le filtre vaut 1).
Les courbes rouge et noire représentent, respectivement, le module et la
partie imaginaire de la transformée de Fourier, la courbe bleue représente le
filtre et la courbe violette est la fonction résultant de l'application du
filtre à la transformée de Fourier, dont on va calculer la transformée de
Fourier inverse.
Quand le filtre vous convient, un clic sur le bouton de fermeture permet de
passer au paramétrage des autres options du calcul de la transformée de
Fourier inverse, grâce à la boîte de dialogue ci-dessous.
La partie supérieure permet d'indiquer la méthode et l'intervalle de calcul
(dans l'espace des k) de la transformée de Fourier inverse, ainsi que
les informations à conserver au cours du calcul.
La partie inférieure permet d'indiquer les paramètres de la fenêtre
utilisée pour le calcul de la transformée de Fourier, afin de pouvoir en
éliminer les effets à l'issue du calcul. Ces paramètres sont mémorisés lors de
la transformation de Fourier et ne sont donc donnés qu'à titre indicatif
ici ; ils peuvent cependant être modifiés, si ces informations ont été
perdues.
Pour le spectre traité, la transformée de Fourier présente essentiellement
un seul pic au voisinage de 2 Å. Il peut être isolé en utilisant comme
filtre une fonction de Hanning avec Rmin = 0,84 Å,
R1 = 1,06 Å, R2 = 1,78 Å
et Rmax = 1,91 Å. Étant donnée la fenêtre utilisée
pour le calcul de la transformée de Fourier, le calcul peut être fait pour
k compris entre 1,5 et 13,5 Å-1, par pas de
0,02 Å-1. La seule information à conserver est le spectre
EXAFS.
À l'issue du calcul, on obtient le spectre EXAFS filtré, pondéré par
k2, dû aux réflecteurs situés vers 2 Å de l'atome
absorbeur, c'est-à-dire le spectre des premiers voisins, les quatre atomes
d'oxygène. C'est ce spectre, représenté ci-dessous, qui va servir de référence
aux modélisations théoriques, dans le cadre d'un modèle de réflexions simple.
La création du modèle demande de connaître le logiciel utilisé pour
réaliser les calculs théoriques et de formuler une hypothèse sur la structure
du composé étudié. LASE étant plus adapté pour travailler avec FEFF, c'est la
réalisation d'un modèle théorique pour ce dernier qui est présentée ici ;
pour la signification précise des différents paramètres, consultez la notice
de FEFF (une version française de la notice de FEFF 6 est disponible sur
demande).
Nous nous limitons ici à l'étude de la contribution de la première
couche de voisins du fer ; cette couche contribue essentiellement
au signal par le terme de réflexion simple. Dans ce cas, seule la
distance entre le fer et l'atome réflecteur intervient ; ce
réflecteur, pour le complexe étudié, est un atome d'oxygène situé à
environ 2 Å du fer. De plus, le nombre d'atomes à une même
distance (ici 4) contribue uniquement par un facteur multiplicatif. Le
modèle structural le plus simple est donc un modèle contenant l'atome
de fer (absorbeur) à l'origine et un atome d'oxygène placé à 2 Å
du fer donc, par exemple, en (2, 0, 0) --- les coordonnées
étant exprimées en angströms dans FEFF.
Pour pouvoir créer ce modèle théorique, il est nécessaire de passer LASE en
mode « Théorie ». Cela se fait en choisissant l'option
>>> Calculs théoriques dans le menu LASE-1 ;
la barre de menu est modifiée en conséquence et devient :
Toutes les options concernant FEFF sont groupées dans le menu FEFF
de cette nouvelle barre de menus. La création pratique du modèle utilisable
par FEFF se fait en trois étapes (correspondant aux trois premières options du
menu) avant d'être enregistrée dans un fichier feff.inp prêt à être
utilisé par FEFF.
L'information fondamentale dans le calcul du modèle, et pour son
utilisation dans LASE ensuite, est la nature de l'atome
absorbeur. Lorsqu'un modèle est créé sans modèle structural dans LASE,
ce qui est le cas ici, il faut donc commencer par préciser cette
information : c'est ce que permet la boîte qui apparaît en tout
premier lieu, dans laquelle on peut entrer le numéro atomique ou le
symbole de l'élément (Fe dans l'exemple traité).
Ensuite apparaît la boîte ci-dessous :
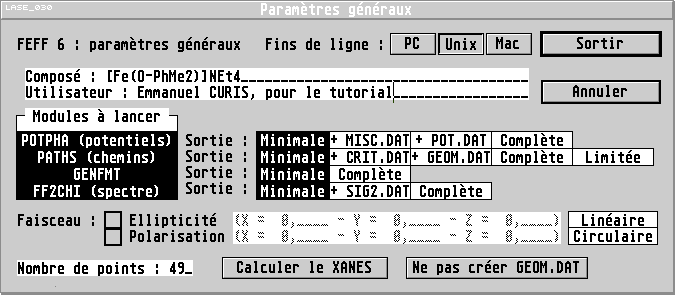
Cette boîte de dialogue permet de régler quelques paramètres
généraux n'ayant pas d'influence directe sur le calcul, ou qui ne sont
pas spécifiquement liés à l'échantillon : nom du composé
modélisé, type de fichier à générer (FEFF est très sensible aux fins
de lignes, un fichier FEFF avec des fins de ligne pour MS-DOS ou
Windows ne sera pas reconnu par la version unix de FEFF, et
réciproquement), nature des informations sauvées par FEFF, faut-il
calculer le X. A. N. E. S.,... Pour davantage
d'informations à ce sujet, voir la
description détaillée. À l'exception du nom du composé et de
l'utilisateur, les paramètres par défaut conviennent (pensez aussi à
choisir le bon type de format de fichier).
Cette boîte de dialogue permet de définir les différents paramètres
influant sur le modèle utilisé par FEFF pour calculer le
potentiel. Dans le cas très simple qui nous intéresse, les seuls
paramètres utiles sont la nature du seuil et la correspondance entre
numéro de potentiel et numéro atomique (lignes Potentiel
x :), sachant que le potentiel 0 correspond
nécessairement à l'atome absorbeur et a donc été défini lors de
l'étape précédente. Le seuil étudié est le seuil K du fer ;
on ne s'intéresse qu'à la contribution de l'oxygène, donc seuls deux
potentiels sont utiles : le potentiel 0 (potentiel du fer)
et le potentiel 1 (potentiel de l'oxygène), ce qui conduit au
paramétrage indiqué sur la figure ci-dessus.
Cette boîte de dialogue permet de définir le modèle structural utilisé par
FEFF pour réaliser le calcul des oscillations EXAFS. Cette structure peut être
indiquée de deux façons :
Pour le modèle choisi, la première méthode de décrire le couple (Fe, O)
étudié est beaucoup plus rapide et plus simple, elle sera donc la seule
envisagée ici.
Il faut, pour décrire le système étudié, créer deux atomes :
un atome de fer de potentiel 0 placé en (0, 0, 0) et un
atome d'oxygène de potentiel 1 placé en (2, 0, 0) ---
toutes les coordonnées sont exprimés en angströms. Pour créer un atome
ex nihilo, cliquez sur le bouton Créer atome. Il
apparaît alors une boîte de dialogue qui permet d'indiquer les
coordonnées, le numéro atomique et le potentiel du nouvel atome. Il
n'est pas nécessaire de créer l'atome absorbeur, car il a déjà été
créé (et placé par défaut à l'origine) lors de l'appel des paramètres
généraux.
Les atomes utilisés dans le modèle, à l'exception de l'absorbeur,
apparaîssent dans le défileur à gauche de la boîte de dialogue ;
les coordonnées et le potentiel de l'atome choisi dans la liste sont
affichés à droite de ce sélecteur (et peuvent y être modifiés). Créer
le modèle de cette façon conduit à la boîte de dialogue telle qu'elle
est affichée ci-dessus.
Elle se fait en choisissant l'option Sauver FEFF.INP du menu
FEFF. Le programme vous permet de sauvegarder le fichier de commandes
FEFF avec n'importe quel nom, ce qui permet de se constituer un ensemble de
fichiers de commande sans trop de problèmes, mais attention : FEFF ne
peut trouver le fichier que s'il s'appelle réellement feff.inp
(attention aux différences entre minuscules et majuscules sous unix !).
Le plus simple est d'utiliser l'option Lancer FEFF du menu
FEFF, en indiquant comme fichier de paramètres le fichier sauvé
précédemment. Cette commande lance FEFF (sans bloquer LASE), à condition que
vous ayez indiqué un chemin d'accès correct à FEFF lors de l'installation de
LASE. Dans le cas contraire, il est nécessaire de lancer FEFF manuellement
dans le dossier contenant le fichier feff.inp à utiliser.
Lorsque le calcul est terminé, les différents fichiers créés par
FEFF se trouvent dans le répertoire contenant le feff.inp
initial ; si le calcul a été lancé depuis LASE, un message vous
avertit qu'il est fini et le fichier chi.dat obtenu est
automatiquement chargé (si le calcul s'est effectué correctement).
Dans un premier temps, la grandeur intéressante est le signal EXAFS
total calculé par FEFF, khiF(k). Ce signal se trouve dans le
fichier chi.dat, qui contient aussi d'autres grandeurs. Si vous avez
utilisé l'option lancer FEFF, ce fichier est chargé automatiquement
en fin de calcul.
Dans le cas contraire, il faut utiliser pour récupérer
khiF(k) les options d'import qui sont accessibles dans le
mode Extraction de LASE. Pour revenir à ce mode, choisir l'option
>>> Extraction dans le menu Lase-2. La barre
de menu disponible au lancement du programme doit réapparaître. Ensuite,
choisir l'option Importer
FEFF dans le menu Fichiers.
Il apparaît alors la boîte de dialogue ci-dessus, qui permet de choisir
quel type de fichier créé par FEFF traiter et quelles informations en
extraire. Dans le cas présent, le type de fichier est chi.dat et la
seule information à conserver est le signal EXAFS.
Tel qu'il a été calculé par FEFF, le signal théorique n'est pas directement
comparable au spectre expérimental, pour deux raisons :
Le premier problème est facilement résoluble : il suffit d'utiliser
l'option Pondérer par k^n du menu EXAFS et d'indiquer la
bonne pondération dans la boîte de dialogue qui apparaît, après avoir choisi
le signal calculé par FEFF comme graphe courant.
Le second problème est plus délicat. En première approche, les effets
mentionnés modifient essentiellement l'amplitude du signal. La comparaison du
signal expérimental et du signal théorique ne peut donc être que
qualitative : a-t-on correctement reproduit la phase des oscillations
expérimentales ?
Pour aller plus loin, il est nécessaire de chercher les valeurs des
différents paramètres cités (distance Fe-O, sFe-O). La façon de
procéder pour obtenir ces valeurs est décrite dans la partie suivante.
À cette étape du traitement, on dispose d'une part du signal EXAFS obtenu
par filtrage du premier pic de la transformée de Fourier des oscillations
expérimentales (ce signal filtré, noté khie(k), étant
interprété comme la contribution au signal total des réflexions simples sur
les atomes les plus proches de l'atome absorbeur) et d'autre part d'un modèle
théorique approximatif pour décrire ce signal : un atome d'oxygène situé
à 2 Å du fer, les deux atomes étant immobiles ; ce modèle permet de
calculer le signal théorique khiF(k). Du fait de la symétrie
sphérique, khiF(k) vaut un quart au signal théorique calculé
pour quatre atomes d'oxygène placés exactement à 2 Å du fer ; de
plus, la prise en compte des vibrations thermiques (supposées faibles)
introduit un terme multiplicatif en
exp(-2s2k2) :
khith(k) = 4exp(-2s2k2) khiF(k). D'après
la théorie de l'EXAFS, dans le cas considéré, on peut écrire que
khiF(k) = f(k)/kR2
sin(2kR + phi(k)) où R est la distance Fe-O,
f(k) est l'amplitude (incluant le terme d'atténuation dû au
libre parcours moyen fini du photoélectron) et phi(k) le déphasage du
signal ; ces deux dernières grandeurs sont calculées par FEFF et
enregistrées dans le fichier feff0001.dat.
Pour améliorer la description des oscillations expérimentales, on est donc
amené à faire varier les valeurs de s et de R. On cherche, en pratique, les
valeurs de s et de R qui minimisent la fonction :
En pratique, on constate que l'énergie de seuil calculée par FEFF ne
reproduit pas exactement l'énergie de seuil réelle, qui n'est pas exactement
non plus l'énergie de seuil E0 choisie lors de l'extraction
des oscillations expérimentales du fait des incertitudes. Cela conduit donc à
une échelle en k légèrement différente pour le spectre expérimental et
le spectre théorique. On est donc amené à faire intervenir un troisième
paramètre, la différence DE0 entre le E0 choisi et le
E0 calculé par FEFF.
Pour pouvoir réaliser cette recherche des valeurs optimales de ces trois
paramètres, il faut indiquer au programme dans quelle mesure ces paramètres
peuvent varier, quelle valeur initiale utiliser, ainsi que les valeurs
d'amplitude et de déphasage à utiliser.
Les amplitudes et déphasages ont été calculés par FEFF pour une
géométrie donnée, ils sont donc, dans LASE, associés au modèle
structural qui a servi à les générer. Ce modèle structural a ici été
créé « au vol » pendant la construction du modèle FEFF ; il constitue
une molécule. Pour accéder à la liste des molécules, il faut
passer en mode « modèle structural » en choisissant l'option
Structure du menu LASE-1. La barre de menus devient alors :
La liste des molécules (c'est-à-dire des modèles structuraux)
s'obtient à partir de l'option Liste des
molécules du menu Structure. Cette option fait
apparaître la fenêtre suivante :
En haut de la fenêtre se trouve la liste des molécules proprement
dite. Ici, une seule molécule existe, celle créée pour le modèle FEFF
et qui porte ce nom. Dans la moitié basse se trouvent les détails des
modèles de la molécule choisie : à gauche, la liste des atomes marqués
comme donnant un seuil d'absorption X (« absorbeur » --- ici, un seul,
créé en même temps que le modèle FEFF : l'atome de fer), à droite les
modèles proposés pour l'absorbeur choisi. Le seul modèle précisé est
celui créé dans la partie précédente, qui ne contient pas les phases
et amplitudes.
Pour charger phases et amplitudes dans un modèle, cliquer sur le
bouton Nouveau. Il apparaît alors la boîte de dialogue suivante :
Cette boîte conditionne l'utilisation des résultats obtenus par
FEFF ; pour l'exemple choisi, les paramètres par défaut conviennent
(plus
d'informations). Dans le sélecteur qui apparaît alors, indiquer le
fichier paths.dat à utiliser.
En voulant construire le modèle, LASE détecte que l'atome absorbeur
est le même que celui déjà en mémoire, qui possède déjà un modèle : il
demande donc si le modèle chargé doit remplacer ou s'ajouter à celui
existant. Comme l'existant ne contient ni phase, ni amplitude, il
n'est pas utile de le garder : on va le remplacer par le modèle
chargé. Les phases et amplitudes sont alors construites, formant des
graphes dans une nouvelle série.
Le modèle étant maintenant complet, il est possible de le
paramétrer. Ce paramétrage comprend deux aspects : les paramètres qui
ne dépendent pas du modèle structural (intervalle à reproduire,
pondération utilisée,... : paramtres généraux) et les autres.
Pour accéder à ces paramètres, il suffit de cliquer sur le bouton
Modifier de la liste des molécules. Il apparaît alors la
fenêtre suivante :
La moitié droite indique les résultats en fin d'ajustement : ils
sont sans intérêt à cette étape de l'analyse. La moitié gauche permet
de définir la façon dont est réalisé l'ajustement, en particulier
l'intervalle à reproduire (de 2 à 12 Å-1 convient ici), les
conditions d'arrêt (celles indiquées par défaut conviennent) et
l'utilisation éventuelle des barres d'erreur. Pour plus
d'informations, voyez
ici.
On peut ensuite modifier les paramètres plus spécifiques, grâce au
bouton Paramètres spécifiques du type de modèle, qui fait
apparaître la boîte de dialogue suivante :
Dans le modèle ajusté, chaque terme de la somme correspond à une
couche dans LASE. La partie supérieure permet de régler les
paramètres les plus généraux, communs à toutes les couches : modèle
pour le libre parcours moyen, pondération utilisée,... La partie
inférieure permet de définir les variables ajustées, couche par
couche. Pour le mode d'emploi détaillé, consultez ce lien.
Dans notre exemple, le modèle comporte une seule couche constituée
de quatre atomes identiques situés à 2 Å de l'absorbeur ;
deux paramètres servent à l'ajustement, la distance R et le facteur de
Debye-Waller s². Cette couche a été créée lors du chargement du
fichier paths.dat, mais (compte-tenu du modèle FEFF utilisé)
avec un seul voisin (N = 1). Avant de lancer l'ajustement, il faut
corriger le nombre de voisins et indiquer 4 : pour cela, cliquer sur
le nom de la variable) modifier dans la liste et changer la valeur
initiale.
Pour démarrer l'ajustement des paramètres structuraux, il faut d'abord
choisir le spectre de référence et le sélectionner comme graphe courant :
il doit être affiché dans la fenêtre principale. Le spectre à utiliser est le
spectre EXAFS obtenu par transformée de Fourier inverse du premier pic de la
transformée de Fourier du spectre expérimental (contribution de la première
couche de voisins au spectre EXAFS).
Ensuite, il faut passer en mode « Ajustements » (la barre de menus
ci-dessous doit apparaître), puis choisir l'option Ajuster le spectre
du menu Couches pour démarrer
l'ajustement .
À l'appel de cette fonction, il apparaît une
fenêtre qui présente les résultats au fur et à mesure de l'ajustement.
Dans cette fenêtre, le spectre de référence est tracé en noir, toujours en
entier ; le spectre obtenu théoriquement par ajustement est tracé en
rouge. La différence entre les deux (résidu) est tracée en bleu. À chaque
itération de l'algorithme de minimisation, ce tracé est mis à jour. La colonne
de gauche donne les valeurs numériques obtenues par l'ajustement :
erreurs et valeurs des paramètres pour chaque couche. Les boutons fléchés
permettent de passer d'une couche à l'autre.
Dans l'exemple choisi, on obtient en fin de minimisation les valeurs suivantes :
Première modélisation de µ0
Première correction à la modélisation de µ0

Seconde correction à la modélisation de µ0
Séparation des contributions
Calcul de la transformée de Fourier

Isolement de la contribution du premier pic

Création du modèle théorique

Paramètres généraux
Potentiels

Structure

Sauvegarde du modèle
Réalisation des calculs
Récupération des résultats

Comparaison avec l'expérience
Recherche des paramètres structuraux

Chargement des déphasages et amplitudes



Paramètres généraux

Paramètres spécifiques

Lancer l'ajustement


| Distance | Facteur de Debye-Waller | Décalage du seuil |
|---|---|---|
| R = 1,88 Å | s2 = 5,99·10-3 Å2 | DE0 = -1,18 eV |
pour une erreur finale pondérée de 4,98·10-2. Cet ajustement est en fait assez mauvais, ce qui peut s'expliquer en particulier par les trois glitches bien visibles dans le spectre brut originel et qui n'ont pas été éliminés.
Afin de comparer différents spectres obtenus soit par l'expérience, soit par le calcul théorique à partir de différents modèles, il est possible dans LASE de superposer différents spectres. Cela se fait en appelant l'option Tracer... du menu Affichage, quand LASE est en mode « Extraction ». Cette option ouvre une fenêtre, similaire à la fenêtre principale, dans laquelle sont dessinés tous les spectres marqués comme visibles.
Pour marquer un spectre comme visible ou invisible, il faut appeler la liste des graphes : tous les spectres cochés sont visibles, et réciproquement ; pour changer l'état visible ou invisible d'un spectre, il suffit de cliquer sur le spectre considéré en maintenant la touche shift enfoncée.
| Présentation du logiciel | Sommaire | Tutorial, partie 2 |