
Ce texte montre, sur quelques exemples, comment créer un modèle structural avec LASE (à partir de coordonnées cristallographiques, par exemple), comment utiliser ce modèle pour créer un modèle utilisable par FEFF et comment confronter les résultats de FEFF, les résultats d'ajustement et la structure tridimensionnelle entrée dans LASE pour faciliter la construction d'un modèle cohérent pour reproduire les spectres expérimentaux.
Cette présentation suppose implicitement que l'on travaille avec une version de LASE autorisant un affichage de la structure tridimensionnelle (compilation avec support Open-GL). Si tel n'est pas le cas, la plupart des opérations décrites ici restent possibles mais deviennent plus délicates et bien moins intuitives.
Ce document contient trois grandes parties :
Pour effectuer cette présentation, l'exemple choisi est le spectre d'absorption des rayons X du cisplatine solide, enregistré au seuil LIII du platine. Le fichiers EXAFS (brut) correspondant se trouve dans l'archive des exemples, sous le nom cispt_exf.txt.
La structure tridimensionnelle peut être définie dans LASE soit directement en entrant les coordonnées, soit en chargeant un fichier qui contient ces coordonnées. Les coordonnées peuvent être soit absolues (dans un repère orthonormé) --- comme on peut les trouver pour une molécule isolée ---, soit fractionnaires --- autorisant l'utilisation directe de données cristallographiques.
LASE peut actuellement charger trois types de fichiers : les fichiers PDB (Protein Data Bank), les fichiers de la Cambridge Crystallographic DataBank et des fichiers en « format libre » en ASCII.
L'unité structurale dans LASE est la molécule, terme à prendre au sens large comme l'ensemble des atomes formant une entité chimique suffisante pour reproduire le spectre d'absorption X. Ce peut donc être aussi bien une vraie molécule, au sens chimique du terme, qu'un lot d'une ou plusieurs mailles d'un cristal (moléculaire, ionique, covalent ou métallique). Il est possible de travailler avec plusieurs molécules simultanément.
Une molécule est constituée d'un ensemble de fragments, correspondant à un groupe d'atomes qu'il est pratique de grouper : une maille dans un ensemble de mailles, un acide aminé dans une chaîne polypeptidique, un ligand dans un complexe,... Elle comprend aussi des atomes absorbeurs qui sont assimilés aux atomes absorbant les photons X.
Les diverses fonctions permettant de construire et modifier le modèle structural sont regroupées dans le mode Structure, correspondant au menu LASE-4 représenté ci-dessous.
Les coordonnées cristallines du cisplatine étant connues, nous allons les utiliser pour générer une structure tridimensionnelle à partir de laquelle le modèle du spectre expérimental pourra être élaboré. Ces coordonnées sont dans le fichier cispt_coord.txt.
Pour charger ce fichier, il faut utiliser l'option fichier ASCII du menu Fichiers (en mode structural : menu LASE-4). La boîte de dialogue suivante apparaît alors.

La partie gauche permet de définir le format du fichier. Ici, le fichier est composé de quatre colonnes. La première contient le symbole chimique de l'élément, les trois suivantes contiennent ses coordonnées. Les lignes de commentaire sont introduites par le symbole # et les paramètres de maille ne sont pas contenus dans le fichier.
La partie droite précise le type de coordonnées contenues dans le fichier. Ici, il s'agit de coordonnées fractionnaires se rapportant à une maille triclinique. Seule l'unité asymétrique est donnée, il faut donc préciser le groupe d'espace (ici P-1) pour générer l'ensemble de la maille. Pour obtenir les coordonnées orthonormées, il est nécessaire de préciser les paramètres de la maille. On demande de ne construire qu'une seule maille (la meille (0,0,0)). La molécule ainsi créée contient donc un seul fragment ; elle s'appelle « Cisplatine ».
Une fois les réglages effectués, cliquez sur Importer, puis choisissez le fichier dans le sélecteur qui apparaît.
Pour afficher la molécule ainsi créée, utilisez l'option Vue en 3D du menu Structure. Il apparaît alors une fenêtre, similaire à celle représentée ci-dessous.

Dans la moitié gauche est affichée la structure tridimensionnelle. La vue peut être modifiée à l'aide des touches du pavé numérique : + et - rapprochent l'observateur ; 1, 9 (resp. 4, 6 et 2, 8) permettent une rotation autour du premier (resp. deuxième et troisième) axe du repère. Pour un contrôle plus fin, ou pour modifier le point visé, il est possible en appuyant sur la touche espace d'appeler la boite de dialogue ci-dessous, qui permet de modifier tous ces paramètres. Dans la figure précédente, les paramètres sont ceux indiqués dans la figure ci-dessous.

Sur cette vue, les atomes sont représentés avec des couleurs conventionnelles : noir pour le carbone, blanc pour l'hydrogène, rouge pour l'oxygène, bleu pour l'azote, vert pour le chlore. Les autres atomes sont représentés en violet, tel le platine ici. En traits fins sont affichés les axes, en traits épais la maille élémentaire (suivant certains angles de vision, ces traits peuvent apparaître colorés, en bleu ciel pour la maille, par exemple).
Dans la moitié droite de la fenêtre, la partie haute permet de modifier certains paramètres d'affichage et la partie basse permet une comparaison des spectres expérimentaux et théoriques associés à la structure étudiée. Ces points seront repris par la suite.
Avant d'aller plus loin, il est intéressant de repérer les divers atomes, les distances et angles entre ces atomes, de façon à contrôler la validité de la structure (pas d'erreur de frappe) et de commencer à distinguer les atomes proches de l'absorbeur ou les atomes presque alignés pouvant contribuer de façon notable par le jeu de la réflexion multiple.
Toutes ces opérations sont réalisables en cliquant sur les atomes affichés dans la vue en trois dimensions. Le type d'information affichée est choisi à l'aide du menu qui apparaît en cliquant avec le bouton du milieu sur la vue en 3 dimensions. Les principales options sont décrites dans le tableau suivant, les autres options seront présentées plus loin.
| Option | Informations fournies |
|---|---|
| Distance entre 2 atomes | Distance entre le premier atome cliqué et le deuxième atome cliqué. |
| Distance (enchaînées) | Distance entre le premier atome cliqué et le deuxième atome cliqué. Ce deuxième atome devient le premier atome, un clic sur un troisième atome donne donc directement la distance entre le deuxième atome et le troisième atome |
| Distance (rayonnante) | Le premier atome cliqué sert de référence, donne ensuite la distance entre l'atome cliqué et cette référence |
| Angle formé par 3 atomes | Si l'on clique sur les atomes 1, 2 puis 3, donne l'angle entre les liaisons 1-2 et 2-3 (angle 123). Cet angle est toujours compris entre 0 et 180°. |
| Angle (enchaînés) | Si l'on clique sur les atomes 1, 2 puis 3, donne l'angle entre les liaisons 1-2 et 2-3 (angle 123). L'atome 2 devient ensuite 1, le 3 devient 2 et un clic sur un quatrième atome donne l'angle 234. |
| Choisir l'atome courant | Un clic sur un atome affiche son type, ses coordonnées, le fragment auquel il appartient. C'est le mode par défaut. L'atome cliqué devient aussi l'atome courant, dont on peut modifier les paramètres (voir plus loin). |
À la simple visualisation de cette structure, on constate que l'on dispose d'une molécule de cisplatine complète, en revanche il ne sera pas possible d'intégrer au modèle des contributions intermoléculaires car l'empilement n'est pas reproduit.
Pour remédier à cela, il est nécessaire de créer de nouvelles mailles de façon à étendre le modèle dans l'espace. Cela peut se faire à l'aide de l'option Créer une nouvelle maille du menu obtenu en cliquant avec le bouton du milieu sur la vue tridimensionnelle. Pour l'exemple choisi, obtenir une molécule complète encadrée par les deux molécules les plus proches (de l'atome de platine considéré) demande d'ajouter les mailles (0, 1, 1), (1, 1, 1), (0, 1, 0) et (0, 0, 1). Il peut être plus rapide de demander, lors de la création de la molécule, directement la création de toutes les mailles entre (0, 0, 0) et (1, 1, 1) --- tout dépend de la molécule et de la structure.
Après cette création de plusieurs mailles, certains atomes peuvent apparaître beaucoup trop loin. S'ils ne pertubent pas les calculs ni le traitement ultérieur, il peut être souhaitable (pour que la vue soit plus claire) de ne pas les utiliser. Deux méthodes permettent cela.
En premier lieu, il est possible d'effacer les atomes superflus. Pour cela, il faut choisir l'option Effacer un atome du menu Opérations sur les atomes qui apparaît en cliquant avec le bouton du milieu sur la vue tridimensionnelle. Ensuite, chaque atome cliqué sera effacé définitivement (après demande de confirmation).
Une solution moins radicale consiste à marquer certains atomes comme invisibles. Par défaut, l'affichage utilise de toute façon tous les atomes. Pour n'afficher que les atomes marqués comme visibles, il faut activer l'option Marqués visibles en haut, à gauche de la fenêtre de la vue tridimensionnelle.
Par défaut, tous les atomes sont marqués comme visibles. Pour sélectionner seulement certains atomes, il faut afficher la liste des atomes en choisissant l'option Liste des atomes dans le menu Structure. On obtient alors la fenêtre de la figure ci-dessous.

Dans cette liste apparaissent tous les atomes actuellement en mémoire (pour toutes les molécules), par ordre de création. L'atome courant est indiqué en surbrillance, pour cet atome sont affichés le numéro atomique, les coordonnées et le rayon, utilisé pour dessiner les sphères (ces informations sont modifiables). Un atome est marqué visible s'il apparaît précédé d'un crochet dans la liste. On peut choisir l'atome courant en cliquant sur l'atome désiré dans la liste. Si l'on appuie sur Shift simultanément, on modifie le caractère visible ou non de l'atome (ce qui n'a d'effet que si le bouton Marqués visibles de la vue tridimensionnelle est activé). Le fonctionnement est donc similaire à celui de la liste des graphes.
Une fois la structure créée et modifiée, il est possible de la sauvegarder pour un usage ultérieur. Pour cela, il faut utiliser le bouton Sauver dans la boîte de dialogue présentant la liste des molécules en mémoire ; la molécule ainsi sauvée est la molécule courante. Pour la recharger, le bouton Charger de cette même fenêtre peut être utilisé.
Le fichier (binaire, donc utilisable uniquement sur le type d'ordinateur sur lequel il a été créé) contient l'ensemble des atomes de la molécule, ainsi que leur agencement en fragments ; les paramètres de maille, s'ils sont connus ; les atomes absorbeurs avec les informations associées, y compris les spectres et les chemins de diffusion (avec phases et amplitudes associées, ainsi que les couches correspondantes dans le modèle d'ajustement - voir plus loin)
Il serait possible de construire un modèle pour FEFF entièrement de la façon présentée dans la première partie de ce tutorial --- méthode qui reste de toute façon indispensable pour paramétrer finement les options de modélisation. Néanmoins, lorsque l'on dispose déjà de la structure tridimensionnelle, LASE propose divers outils qui facilitent la création de ce modèle.
L'atome qui est considéré comme celui excité par les photons X absorbés (« atome absorbeur »)joue un rôle considérable, aussi LASE permet de définir certains atomes comme des atomes absorbeurs. Chaque molécule peut contenir un nombre quelconque d'atomes absorbeurs. Si un atome est défini comme absorbeur, LASE sait qu'il sert de point central autour duquel sera construit un modèle de spectre d'absorption (avec FEFF en particulier). Il est alors possible de lui associer diverses informations à propos de ce modèle (chemins de réflexion trouvés par FEFF, spectre expérimental de référence,...) qui facilitent l'analyse des résultats obtenus à l'issue d'un ajustement. Il est donc préférable d'indiquer, dans la structure, quels atomes peuvent être considérés comme des atomes absorbeurs.
La façon la plus simple d'indiquer qu'un atome est un absorbeur est de choisir l'option Choisir l'atome courant dans le menu Opérations sur les atomes. Dans ce mode, un clic simple sur un atome le définit comme atome courant. En revanche, si l'on appuie sur la touche Ctrl en même temps que l'on clique, on indique que l'atome choisit doit être un atome absorbeur. Après demande de confirmation, il apparaît alors la boîte de dialogue ci-dessous, qui permet de préciser les paramètres de cet atome absorbeur.

Il est en particulier possible de nommer cet absorbeur et de lui associer un spectre de référence : spectre EXAFS expérimental observé au seuil de cet atome. Ici, on peut lui assigner le spectre EXAFS brut présenté au début de ce document.
L'atome absorbeur actif est affiché en orange dans la vue tridimensionnelle. Si le bouton Expérience est activé et si un spectre de référence a été associé à l'absorbeur, il est affiché dans la partie inférieure droite de la fenêtre de visualisation tridimensionnelle (figure ci-dessous).
Pour construire un modèle FEFF, il faut
essentiellement définir les potentiels (correspondance entre le numéro
du potentiel et le numéro atomique) et la disposition des atomes dans
l'espace. Le principe de l'interface proposée dans LASE
pour réaliser ces définitions a été présenté dans cette partie et ne sera pas repris
ici.
Lorsqu'un ou plusieurs atomes absorbeurs ont été définis, ces deux
étapes sont cependant grandement accélérées. En effet, l'atome
absorbeur a nécessairement le potentiel 0 et sert de base à la
construction du modèle. Aussi, lorsqu'un atome absorbeur existe,
LASE peut l'utiliser directement pour créer le modèle,
ce qui évite d'indiquer sa nature lors de la première étape de
construction du modèle.
Une fois l'atome absorbeur choisi, le modèle est le plus souvent
construit en intégrant tous les atomes qui se trouvent à moins d'une
certaine distance de l'atome absorbeur, typiquement 5 Å. L'atome
absorbeur mais aussi son environnement ayant été définis dans le
modèle structural, LASE peut automatiquement construire
cette liste d'atomes à partir de la structure tridimensionnelle. Pour
cela, il suffit de cliquer sur le bouton Tout choisir de la
boîte de dialogue permettant de définir la structure du modèle
FEFF. Si un atome absorbeur a été défini, il apparaît
alors la boîte de dialogue ci-dessous, qui permet de contrôler la
façon dont sont ajoutés les atomes au modèle.
Ce contrôle permet d'ajouter soit tous les atomes de numéro
atomique donné, soit tous les atomes à moins d'une certaine distance
de l'atome absorbeur. Ces atomes appartiennent nécessairement à la
même molécule que l'atome absorbeur. Au cours de cet ajout d'atomes,
LASE s'occupe de créer de nouveaux potentiels (dans la
limite des huit autorisés par FEFF) à chaque fois qu'un
nouveau type d'atome est rencontré.
Pour l'exemple considéré, choisir tous les atomes à moins de
5 Å de l'atome de platine choisi comme absorbeur conduit à
ajouter 19 atomes, correspondant à trois molécules complètes
(appartenant à une même colonne) et des atomes des molécules des
colonnes voisines.
Les atomes effectivement choisis pour le modèle FEFF peuvent être
mis en évidence dans la vue tridimensionnelle en activant le bouton
Modèle FEFF : dans ce cas, seuls les atomes
effectivement utilisés sont affichés.
Attention ! LASE ne contrôle
pas que la sphère indiquée est cohérente avec la structure
indiquée. Ainsi, si l'on choisit un atome en bord de maille avec une
seule maille définie, la sphère centrée sur cet atome peut inclure des
mailles voisines qui sont absents du modèle. Comme, fondamentalement,
la structure indiquée ou étudiée n'est pas forcément cristalline,
LASE n'a aucun moyen sûr de contrôler que la sphère
définie est bien « homogènement pleine », c'est à l'utilisateur de
s'en assurer. Voir les atomes inclus au modèle FEFF
permet souvent d'indiquer un problème, si la région occupée autour de
l'absorbeur laisse manifestement un grand domaine spatial vide dans un
angle solide donné.
La sélection automatique permet de « dégrossir » la construction du
modèle, mais il est parfois nécessaire d'affiner le modèle en ajoutant
ou en ôtant manuellement certains atomes.
S'il est possible de le faire dans la boîte de dialogue définissant
la structure pour le modèle FEFF, à partir de la liste
des atomes, il est aussi faisable dans LASE d'indiquer
directement sur la vue tridimensionnelle les atomes à ajouter ou
enlever.
Pour cela, il faut sélectionner dans le menu Opérations sur les
atomes apparaissant lorsque l'on clique avec le bouton du milieu
sur la vue tridimensionnelle le mode Ajouter des atomes au
modèle (pour ajouter des atomes) ou le mode Enlever des
atomes au modèle (pour enlever des atomes). Lorsque l'on est dans
ce mode, tout atome cliqué est ajouté ou enlevé du modèle.
Une fois le modèle structural achevé, le fichier feff.inp
est créé suivant la méthode usuelle, puis le calcul lancé
classiquement.
Dans le modèle d'une décomposition en chemins des oscillations
EXAFS, les oscillations observées correspondent à la somme des
contributions des divers chemins. Pour obtenir les paramètres
structuraux par ajustement, il faut donc créer une
« couche » par chemin trouvé dans le modèle
FEFF (il y a équivalence entre couche et chemin pour
les chemins de réflexion simple), chaque couche correspondant donc à
un terme de la somme.
S'il est envisageable de construire manuellement, à partir des
résultats de FEFF, les couches utilisées pour
l'ajustement (en précisant pour chacune le déphasage et l'amplitude,
ainsi que les valeurs initiales des paramètres), en utilisant la
méthode décrite dans la première
partie, la tache devient vite fastidieuse.
LASE permet donc de construire ce jeu de chemins
directement à partir des fichiers de résultats créés par
FEFF : fichier paths.dat décrivant les
chemins trouvés et fichiers feffxxxx.dat décrivant,
chemin par chemin, les déphasages et amplitudes.
Pour réaliser cette génération automatique des chemins, il faut
commencer par appeler la liste des molécules
(menu Structure), rappelée
ci-dessous.
Le bouton Nouveau permet de créer un nouveau modèle pour
l'absorbeur actif --- ou, si un modèle de même type existe déjà, de le
remplacer. Un clic sur ce bouton appelle la boîte de dialogue
suivante :
qui permet de contrôler la façon dont sont créés les chemins de
diffusion utilisés pour l'ajustement. Par défaut, tous les chemins
trouvés par FEFF sont utilisés, avec une dégénérescence
(« nombre de voisins ») fixée et une longueur de chemin variable,
le terme de libre-parcours moyen étant inclus à l'amplitude de chaque
chemin. Une fois ce paramétrage effectué, un clic sur
Construire appelle le sélecteur de fichier pour indiquer la
localisation du fichier paths.dat. Les fichiers
feffxxxx.dat doivent se trouver au même endroit pour
pouvoir générer les déphasages et amplitudes de ces chemins.
On obtient, à l'issue du chargement, un jeu de couches et de
chemins prêts pour lancer un ajustement (à condition de régler les
paramètres généraux).
Avec un modèle structural un tantinet complexe, le nombre de
chemins de réflexion trouvés par FEFF devient très
rapidement important - en particulier lorsque les symétries autour de
l'absorbeur ne sont pas conservées à cause de la « trop grande
précision » des coordonnées cristallines. Si l'on autorise, pour
chaque chemin, ne serait-ce que la longueur de chemin et le facteur de
Debye-Waller à varier, on se retrouve très vite avec un nombre
conséquent de variables, ce qui permet bien sûr une très bonne
reproduction du signal expérimental, mais pose aussi des problèmes de
validité physique des résultats ainsi obtenus et induit souvent une
convergence vers un minimum physiquement absurde à cause des fortes
corrélations entre variables.
Par suite, il est nécessaire de réduire ce nombre de chemins et de
vérifier la validité physique du minimum obtenu. LASE
propose quelques options en ce sens, que nous présentons ici, mais le
choix d'une heuristique pour ajouter ou ôter des chemins est en
dernier recours du ressort de l'utilisateur et reste la tache la plus
délicate de la construction d'un modèle.
Considérons les chemins trouvés par FEFF pour le
modèle du cisplatine indiqué. FEFF trouve
25 chemins, en se limitant aux trois molécules d'une même colonne
(voir cette figure). Parmi ces 25 chemins,
les chemins 3 et 4, par exemple, correspondent tous les deux à une
réflexion simple sur l'atome de chlore lié à l'atome de platine
absorbant les rayons X. FEFF trouve deux chemins
car les coordonnées cristallines indiquées conduisent à deux distances
platine-chlore, très proches, de 2,2844 Å et 2,2328 Å. Si
l'on raisonne sur des atomes fixes, il est alors légitime de
distinguer ces deux chemins ; toutefois, du fait de l'agitation
thermique, ces deux atomes de chlore sont en fait indiscernables par
EXAFS : il est donc plus correct de n'utiliser qu'un seul chemin
de réflexion, mais avec une dégénérescence double.
En procédant à une analyse similaire sur l'ensemble des chemins, on
arrive souvent à réduire de façon très importante le nombre de chemins
à prendre en compte dans le modèle.
Pour faciliter cette analyse, LASE permet d'afficher
chacun des chemins de diffusion sur la vue tridimensionnelle. En
activant l'option Chemins dans la boîte en haut à gauche de
la fenêtre, le chemin de réflexion actif est dessiné en trait vert
épais sur la vue tridimensionnelle. Pour le rendre plus visible, il
peut être souhaitable de diminuer la taille des sphères représentant
les atomes, souvent jointives. Pour cela, cliquer sur
Atomes : Sphères, taille réelle et, dans le menu qui
apparaît, choisir Sphères, échelle 1:3. On obtient ainsi une
représentation similaire à celle de la figure ci-dessous.
Ici, le chemin choisi part de l'absorbeur, passe par l'un des
atomes d'azote puis l'autre avant de revenir à l'atome absorbeur. On
peut choisir le chemin affiché avec le défileur en bas de la boîte de
dialogue : les flèches passent au chemin précédent ou
suivant ; un clic au centre (où est affiché le nom du chemin)
affiche la liste de tous les chemins pour en choisir un directement.
En affichant successivement tous les chemins, il est possible de
repérer assez rapidement les chemins identiques par symétrie, ce qui
permet de réduire le nombre total de chemins. Pour l'exemple choisi,
on passe ainsi de 25 chemins à 13 chemins (en regroupant au
maximum).
En pratique, les chemins « en double » sont éliminés en
désactivant le bouton Utiliser pour la couche associée (on
n'utilise pas la couche lors de l'ajustement) et --- pour ne pas
perdre d'atomes lors de cette condensation --- en modifiant le nombre
de voisins pour les couches concernées. Par exemple, dans le modèle
ci-dessus, les chemins 3 et 4 sont assimilables (réflexion simple
sur un atome de chlore lié à l'atome absorbeur). On désactivera donc
la couche associée au chemin 4 et on indiquera 2 voisins
pour la couche associée au chemin 3 (les couches originelles
comptant chacune 1 voisin).
Cette visualisation des chemins permet aussi d'éliminer des chemins
sur d'autres critères structuraux (réflexion sur plus de deux
atomes légers sans alignement particulier, par exemple) en les
repérant rapidement sur la vue tridimensionnelle.
Une fois ces opérations préalables effectuées, l'ajustement peut
être lancé selon la procédure habituelle.
À l'issue d'un ajustement, on peut très bien obtenir une bonne
reproduction du spectre expérimental mais des paramètres physiques
absurdes --- en particulier lorsque un grand nombre de paramètres a
été utilisé pour réaliser cet ajustement.
L'incohérence des paramètres obtenus peut porter sur divers
aspects, plus ou moins faciles à repérer. LASE peut
signaler les plus manifestes. Pour cela, il faut afficher la fenêtre
des paramètres généraux d'un
modèle puis, dans la partie droite, cliquer sur le bouton de
validité. Il apparaît alors un menu, qui propose en particulier de
vérifier le sens physique du résultat, ainsi que la vérification des
valeurs obtenues par rapport aux bornes de variation imposées
(Bornes atteintes ?).
Lorsque l'option de sens physique du résultat est appelée, LASE signale :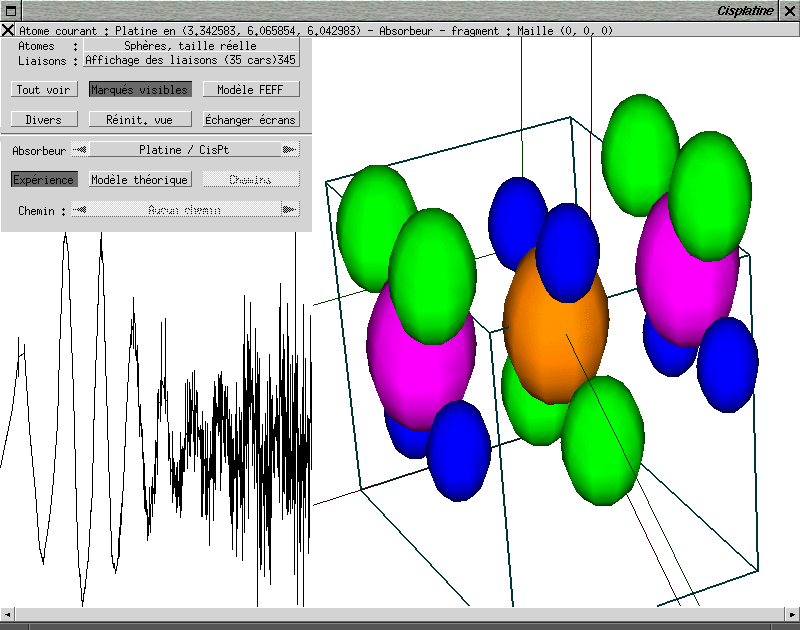
Création du modèle FEFF

Affiner manuellement le modèle FEFF
Utilisation des résultats pour l'ajustement


Validation des ajustements
Utilisation des symétries

Validité des paramètres obtenus
et propose d'éliminer du modèle la couche incriminée.
| Tutorial, partie 2 | Sommaire |